Wochenrückblick KW 45
Gentherapie gegen Immunkrankheit wirksam
Wissenschaftler der Medizinischen Hochschule Hannover (MHH) haben erstmals mittels einer Stammzellen-Gentherapie Kindererfolgreich behandeln können, die an der Erbkrankheit Wiskott-Aldrich-Syndrom litten.
Den Medizinern ist es gelungen, den zugrunde liegenden Gendefekt zu beheben und die Symptome der Immunkrankheit zu beseitigen.
Acht von zehn der jungen Patienten führten auch bis zu vier Jahre nach dem Eingriff ein normales Leben. Erste Ergebnisse der beiden am längsten erfolgreich behandelten Patienten sind in der Fachzeitschrift New England Journal of Medicine (11. November 2010, Bd. 363, S.1918) veröffentlicht. Das Wiskott-Aldrich-Syndrom ist ein Immundefekt, der durch eine Mutation in dem Gen namens WAS ausgelöst wird. Es ist für die Reifung und Aktivierung der Immunabwehr, etwa der weißen Blutkörperchen und der Blutplättchen zuständig. Zu den Symptomen des Wiskott-Aldrich-Syndroms zählen unter anderem schwere wiederkehrende Infektionen wie Lungenentzündungen. Bislang konnten Kinder nur mit Hilfe einer Transplantation von Blutstammzellen eines Spenders gerettet werden. Mit diesem Verfahren sind aber häufig schwere Nebenwirkungen verbunden.

| Mehr auf biotechnologie.de |
News: Gentherapie für die Netzhaut: Es werde Licht Wochenrückblick: Biotechnologen konstruieren maßgeschneiderte Genfähren |
Dagegen sei die Gentherapie weniger belastend als die Transplantation von Blutstammzellen, sagte der wissenschaftliche Mitarbeiter im Rahmen dieser Gentherapiestudie, Kaan Boztug. Die Mediziner hatten für die Therapie ein gesundes WAS-Gen mittels viralem Gentaxi in Blutstammzellen eingeschleust. “Die Korrektur zeigt sich bei acht von zehn Kindern in allen Blutzellen stabil“, berichten die Gentherapeuten aus Hannover. Ein Patient konnte jedoch nicht genug korrigierte Zellen erhalten, ein weiterer entwickelte als Nebenwirkung eine Leukämie. Christoph Klein, Direktor der Klinik für Pädiatrische Hämatologie und Onkologie an der MHH und Leibniz-Preisträger 2010 sagte, neue Therapien würden immer auch das Risiko unerwünschter Nebenwirkungen bergen, diese ließen sich leider auch in der aktuellen Studie nicht vermeiden“. Wenn alles gut läuft, werde eine derartige Gentherapie einmal routinemäßig angewandt und Patienten bleibe vielleicht sogar die lebenslange Einnahme von Medikamenten erspart, so die Mediziner.
Unverträgliche Genvarianten bei Pflanzen aufgespürt
Kölner Entwicklungsgenetiker sind einem Mechanismus auf die Spur gekommen, der bei Pflanzen zur Entstehung von neuen Arten führt.
Dazu haben sie in Pflanzen der Art Arabidopsis thaliana aus verschiedenen Erdteilen nach Genvarianten gefahndet, die sich stark auseinanderentwickelt haben und sich deshalb nicht mehr miteinander "vertragen". Die Forscher berichten im Fachmagazin Nature Genetics (2010, Online-Vorabveröffentlichung) über ein Gen namens SRF3, das eine sogenannte Inkompatibilität auslöst.

Individuen derselben Pflanzenart lassen sich meist problemlos kreuzen, sie bringen Nachkommen hervor, die sich normal entwickeln. Manche Genvarianten vertragen sich jedoch nicht mehr mit denen eines anderen Individuums. Denn durch die natürliche Selektion in den verschiedenen Lebensräumen der Welt können sich Genversionen herausbilden, die nicht mehr zu denen von Pflanzen an anderen Standorten passen. Auf diese Weise können sich neue Arten herausbilden. Pflanzen, die solche unverträglichen Genversionen enthalten, sind häufig steril und wachsen langsam. Züchtungsforscher nennen sie inkompatible Hybride. Trotzdem verschwinden diese Genvarianten nicht einfach wieder aus der Natur. Das legt die Vermutung nahe, dass sie für ihre Herkunftspflanzen von Vorteil sind und sich erst nach der Kreuzung von ihrer schlechten Seite zeigen. Forscher um Matthieu Reymond und Rubén Alcázar vom Max-Planck-Institut für Pflanzenzüchtungsforschung haben nun inkompatible Hybride untersucht, wie sie bei der Kreuzung von Arabidopsis-Varianten verschiedener geographischer Herkunft entstehen. Kreuzt man den europäischen Ökotyp Ler mit den asiatischen Ökotypen Kas-2 oder Kond, erhält man inkompatible Hybride, die bei Temperaturen unter 14 Grad Celsius nur eine Zwergform ausbilden.
| Mehr zum Thema auf biotechnologie.de |
News: Enormes Tempo der Evolution überrascht Pflanzenforscher News: Zusammenspiel der Hormone im Pflanzenspross aufgeklärt |
Bei höheren Temperaturen wachsen sie dagegen normal. Durch molekulare Analysen haben die Kölner Wissenschaftler die verantwortliche Genvariante für diesen Effekt aufgespürt. Das Allel lässt ein Protein namens SRF3 entstehen, das eine stärkere Immunantwort in der Pflanze Gang setzt. Die Kölner Wissenschaftler nehmen an, dass die Ökotypen der Ackerschmalwand über die Zeit hinweg verschiedene genetische Änderungen angesammelt haben. Nach Ansicht der Forscher konnten sich die inkompatiblen Allele vermutlich in Asien ausbreiten, weil sie den dortigen Populationen zu einer optimalen Immunantwort verhelfen. Die desaströse Überaktivität des Immunsystems erzeugen sie erst bei der Kreuzung mit dem Ler-Ökotyp, dem sie aber in der Natur wegen der großen räumlichen Distanz vermutlich nie begegnen werden. Für die schädliche Kombination gibt es deshalb keinen Selektionsdruck.
Membranprotein-Chip testet tausende Wirkstoffe gleichzeitig
Forscher aus Frankfurt und München haben einen Membranprotein-Chip entwickelt, mit dem sich der Transport von mehreren tausend Wirkstoffen gleichzeitig testen lässt.
Dazu hat das Team um Robert Tampé an der Goethe-Universität gemeinsam mit Forschern vom Walter-Schottky-Institut der Technischen Universität München einen Chip mit einer nanostrukturierten Oberfläche konstruiert.
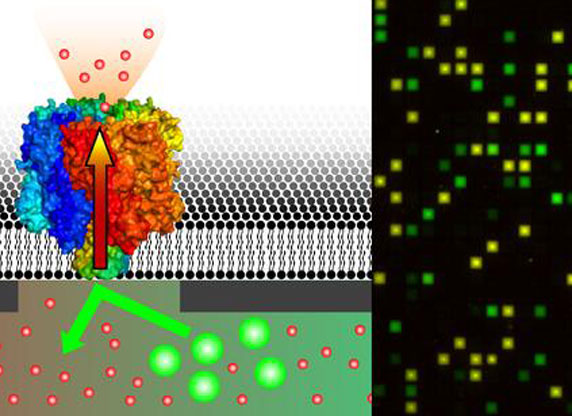
Wie die Wissenschaftler in der Fachzeitschrift Nano Letters (2010, Online-Vorabveröffentlichung) berichten, werden die Membranproteine auf einer Chipoberfläche aufgebracht, auf der sich fast 50.000 Nanoporen befinden. Rezeptoren, Kanäle und Transporter in Zellmembranen zählen zu den wichtigsten Zielmolekülen der Pharmaindustrie. Die Suche nach neuen Wirkstoffen unter Millionen chemisch und strukturell ähnlichen Verbindungen gleicht der Suche nach der Nadel im Heuhaufen.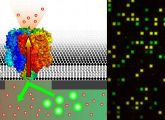
| Mehr auf biotechnologie.de |
Menschen: Frank Bier: Labor im Zwergenmaßstab |
Benötigt werden analytische Techniken, mit denen sich mehrere tausend potenzielle Wirkstoffe parallel an Proteinen testen lassen. Der neue Chip eignet sich zur automatisierbaren Hochdurchsatz-Untersuchung von Wirkstoffen an hochempfindlichen Membranproteinen. Die Verwendung der nanostrukturierten Oberfläche hat den Vorteil, dass die Membranproteine innerhalb von frei tragenden Lipidmembranen auf den winzigen Poren in ihrer natürlichen Struktur und somit auch in ihrer Funktion erhalten bleiben. Auf organische Lösungsmittel in der Lipidmembran, welche ebenfalls die Struktur der Membranproteine zerstören können, kann verzichtet werden. Wie ein Wirkstoff beispielsweise den Transport von Stoffen ins Zellinnere beeinflusst, prüfen die Wissenschaftler mithilfe der Fluoreszenzmikroskopie. Aufgrund der parallelen Architektur des Systems ist eine Vielzahl von gleichzeitigen Messungen möglich.
Micromet: Aktienverkauf spült 70 Millionen US-Dollar in die Kasse
Das deutsch-amerikanische Biotech-Unternehmen Micromet hat durch den Verkauf von Aktien insgesamt 72,3 Millionen US-Dollar eingenommen.
Der an der US-Technologiebörse Nasdaq notierte Antikörperspezialist verkaufte 9,9 Millionen Aktien zum Stückpreis von 7,30 US-Dollar. Micromet war aber auch vor der Kapitalerhöhung schon gut finanziert. Ende September belief sich der Barbestand des Medikamentenentwicklers, der seinen Hauptsitz in den USA hat, aber alle Forschungsaktivitäten in München durchführt, bereits auf 164 Millionen US-Dollar. Rechnet man die aktuelle Finanzspritze hinzu, übersteigen die liquiden Mittel damit die 200 Millionen US-Dollar-Grenze. Die wiederholte Aktienausgabe erlaubt es der Firma, die eigenen Entwicklungsprogramme auszudehnen. Zwar entwickelt Micromet auch zusammen mit Pharmapartnern Antikörperkandidaten in der klinischen Phase, die größte Kursphantasie steckt jedoch in Blinatumomab. Der bispezifische Antikörper bindet sowohl an den CD3- als auch an den CD19-Rezeptor und bringt damit T-Zellen als auch B-Zellen des Immunsystems in direkten Kontakt.
| Mehr zum Thema auf biotechnologie.de |
Wochenrückblick: Micromet holt sich 70 Millionen US-Dollar am Kapitalmarkt Wirtschaft: Micromet weckt Hoffnungen bei Pharmaindustrie |
Dieser Mechanismus soll helfen, eine schwer therapierbare Form von Blutkrebs, der sogenannten akuten lymphatischen Leukämie (ALL), zu heilen. Durch die Bindung an seine Zielproteine führt der Antikörper zu einer Kopplung entarteter B-Zellen an eine T-Zelle des Immunsystems. Diese wird somit aktiviert und löst den programmiertern Zelltod der B-Zelle aus. Im September startete Micromet eine Phase II-Studie mit 130 Patienten in Europa, deren Ergebnisse bereits für eine Zulassung verwendet werden können. In einer weiteren, gerade begonnenen klinischen Studie soll getestet werden, ob der Antikörper auch Patienten mit wiederkehrender ALL helfen kann. Das wäre für das Unternehmen eine interessante Indikationserweiterung. Zudem sollen zwei neue Bite-Antikörper (MT111 und MT112) in die Klinik gebracht werden, deren Rechte bei der AstraZeneca-Tochter MedImmune beziehungsweise der deutschen Bayer Schering AG liegen.
Neue Sequenziermethode verbessert Diagnose von Gendefekten
Forscher des Helmholtz Zentrums München haben mit einer neuen Sequenziermethode im Genom eines einzelnen Patienten die Ursache seiner Stoffwechselerkrankung identifiziert.
Mithilfe der sogenannten Exom-Sequenzierung haben die Forscher um Holger Prokisch und Thomas Meitinger einen Gendefekt aufgespürt, der die molekularen Abläufe der Atmungskette in den Mitochondrien behindert. Wie die Forscher in Nature Genetics (7. November 2010, Online-Vorabveröffentlichung) berichten, verbessert die neue Methode die molekularen Diagnose deutlich und bietet möglicherweise gezielte Therapieansätze für die Patienten. Bei der Exom-Sequenzierung wird nur jener Teil der kompletten Erbinformation entziffert, der tatsächlich für Eiweiße oder funktionelle Genprodukte kodiert. Das sind circa 1,5 Prozent der gesamten DNA. Die Analyse des Patienten-Exoms offenbarte den Sequenzierspezialisten eine Mutation im Gen namens ACAD9. Sie verhindert, dass der sogenannte „Mitochondriale Komplex I“ in den Zellkraftwerken arbeiten kann.
| Mehr zum Thema auf biotechnologie.de |
News: 1000-Genome-Projekt legt menschliche Vielfalt offen Dossier: Jubiläum: Zehn Jahre Humangenom |
In Organen mit hohem Energieverbrauch wie Gehirn, Herz oder Auge werden bei einem solchen Defekt die Veränderungen besonders spürbar. Fehlfunktionen von Mitochondrien werden auch bei der Entstehung der Parkinson-Erkrankung und des Diabetes beobachtet. Symptome dieser Erkrankungen treten auch bei Patienten mit Komplex I-Störungen auf. Das besondere an der vorgelegten Studie: Die Genomanalyse eines einzigen Patienten mit einer seltenen Erkrankung reichte den Forschern, um AKAD9 als Risikofaktor zu identifizieren. Bislang wurde dieses Gen mit dem Fettstoffwechsel in Zusammenhang gebracht. „Wir möchten diese Erkenntnisse nutzen, um künftig Patienten, die an mitochondrialen Erkrankungen leiden, eine konkrete molekulare Diagnose stellen zu können“, sagt Prokisch. Denn je früher die Diagnose gestellt wird, desto schneller können Therapiemaßnahmen getroffen werden. Dies ist bei den jetzt gefundenen Mutationen im ACAD9 bereits möglich: Hier kann gezielt mit der Therapie mit Riboflavin begonnen werden. Generell wird die Methode der Exom-Sequenzierung es ermöglichen, bisher nicht identifizierte Mutationen bei seltenen Erkrankungen zu diagnostizieren, hoffen die Wissenschaftler.
Kleine Salmonellen-RNA drosselt Produktion von zehn Eiweißen
Wird die Zellhülle von Salmonellen beschädigt, so stoppen die Mikroben schlagartig die Produktion einer Reihe von Eiweißen.
Ein kleines RNA-Molekül spielt bei diesem Produktionsstopp eine Schlüsselrolle. Das haben Forscher vom Institut für Molekulare Infektionsbiologie der Universität Würzburg herausgefunden. Das Team um Jörg Vogel berichtet im Fachjournal PNAS (8. November 2010, Online-Vorabveröffentlichung). 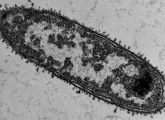
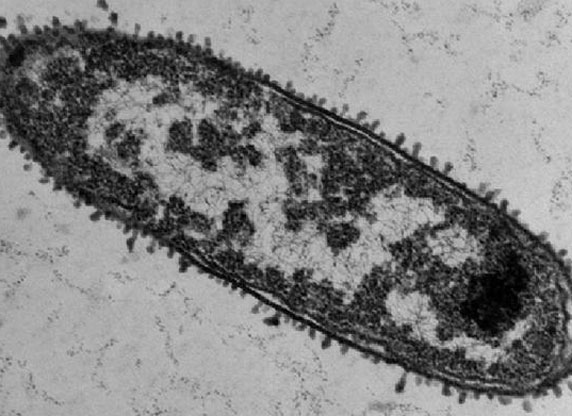
Salmonellen sind Bakterien, die beim Menschen heftigen Durchfall auslösen können. Wenn sie sich im Verdauungstrakt ausbreiten, bleiben sie nicht unbehelligt: Das Immunsystem attackiert die Eindringlinge unter anderem mit Peptiden. Das sind kleine Eiweißmoleküle, die Löcher in die äußere Hülle der Bakterien reißen. Sobald ihre Hülle beschädigt ist, reagieren die Salmonellen ihrerseits: Unter anderem produzieren sie dann ein kleines RNA-Molekül namens RybB, das in der Bakterienzelle schlagartig die Synthese von etwa zehn Proteinen unterbindet. Dabei handelt es sich allesamt um Proteine, die für einen Einsatz in der Außenhülle der Bakterien vorgesehen sind. „Die Salmonellen helfen sich damit ganz schnell. Weil ihre äußere Membran löchrig ist, würden die Proteine dort keinen Halt finden und nicht funktionieren“, erklärt Mitautor Kai Papenfort. Letztlich verhindert das kleine RNA-Molekül so eine Verschwendung von Protein-Ressourcen. Die Forscher gingen der Frage nach, wie es die kleine RNA schafft, auf einen Schlag die Produktion gleich mehrerer Proteine zu regulieren.
| Mehr auf biotechnologie.de |
News: Erster Kontakt: Wie Neugeborene eine gesunde Darmflora aufbauen |
„Der Anfangsbereich des kleinen RNA-Moleküls bindet die Transkripte, also die Vorstufen all dieser Proteine“, sagt Jörg Vogel. „Sobald das geschehen ist, stoppt die Produktion.“ Zum Beweis übertrugen die Forscher den Anfangsbereich auf andere RNA-Moleküle. Auch diese brachten daraufhin die Herstellung der zehn Proteine ins Stocken. Erstmals haben die Würzburger Forscher damit gezeigt: Auch kleine RNA-Moleküle besitzen klar abgrenzbare Bereiche, denen sich eine regulierende Funktion zuweisen lässt. Bislang war das nur für Proteine bekannt, nicht aber für „einfachere“ Moleküle wie RNA. „Auch RNA besteht aus funktionellen Stücken, die sich nach dem Baukastenprinzip neu anordnen lassen“, so Vogel. Da der neue entdeckte Mechanismus offenbar auch bei anderen krankheitserregenden Bakterien vorkommt, könnte sich der Anfangsbereich der RybB-sRNA als potenzieller Angriffspunkt für neue Medikamente erweisen.